Ostatnia aktualizacja: 09/10/2018, Dr. Miguel B. Royo Salvador, Arztnummer: 10389. Neurochirurga i neurologa.
Choroba Filum obejmuje choroby konwencjonalne takie jak: Syndrom Arnolda Chiari I, Idiopatyczna jamistość rdzenia y Skolioza idiopatyczna, Płaskopodstawie (Platibasia), Wgłobienie obrotnika do jamy czaszki (Impresión Basilar), Odchylenie kręgu obrotowego (Retroceso Odontoideo), Wygięcie pnia mózgu (Angulación del tronco cerebral), wpływając tym samym na pojawienie się Dyskopatii (wypuklin i przepuklin dysku), Zwężenia kanału kręgowego,, Moczenia nocnego i innych schorzeń.
Choroba Filum daje objawy głównie ze strony układu nerwowego, głowy i kręgosłupa, są to: bóle głowy, nudności, wymioty, dysfagia, zawroty głowy, zaburzenia pamięci, bóle w odcinku szyjnym, bóle w odcinku piersiowym, bóle w odcinku lędźwiowym, parestezje, zaburzenia wrażliwości, bóle kończyn, brak siły w kończynach, zaburzenia równowagi, bezsenność, zaburzenia chodu i wiele innych.
Choroba Filum zgodnie z naszymi badaniami naukowymi jest konsekwencją trakcji całego układu nerwowego: rdzenia kręgowego, pnia mózgu, móżdżku i mózgu, wywołanej więzadłem filum terminale, które jest anormalne.
Choroba Filum jest najprawdopodobniej dziedziczną chorobą, na którą cierpi co piąta osoba, wystarczająco dużo, aby zastosować leczenie.
Nasz Instytut Chiari & Siringomielia & Escoliosis de Barcelona (ICSEB) podczas swoich badań naukowych opracował metodę Filum System®, która pozwala na dokładne zdiagnozowanie Choroby Filum i jej licznych konsekwencji, stosując odpowiednie leczenie przy użyciu minimalnie inwazyjnych technik chirurgicznych.
Dzięki tej metodzie możliwe jest naprawienie odwracalnych uszkodzeń układu nerwowego i w niektórych przypadkach uzyskanie korzystnych zmian anatomicznych (zanik jamistości rdzenia, podwyższenie migdałków móżdżku, wyprostowanie kręgosłupa i zmniejszenie dyskopatii).
Leczenie może zostać zastosowane u dzieci, dorosłych i osób starszych, ponieważ rekonwalescencja jest szybka i łatwa. (Wyniki leczenia Choroby.)
Prowadzimy badania nad Chorobą Filum od 1975 roku i znajdujemy się w czołówce. ICSEB jest jedynym centrum na świecie, gdzie poza ciągłymi badaniami naukowymi, diagnozujemy i stosujemy metodę Filum System®, wyłączną metodę opisaną przez nas w celu badania rozwoju Choroby Filum, która pozwala układowi nerwowemu na naprawę szkód spowodowanych przez chorobę.
Bardzo często zdarza się, że wyraźnie zdefiniowana Choroba Filum mylona jest z innymi pokrewnymi chorobami o podobnym mechanizmie. Ta sekcja odnośni się do tych chorób, z których część powiązana jest z Chorobą Filum oraz wyjaśnia nowe koncepty syndromu Nerwowo-Czaszkowo-Kręgowego (S. NCV), Choroby Filum (EF) i FILUM SYSTEM® (FS®).
Definicje, nomenklatura i opis konceptów, terminów i chorób, które w dalszej części będą się często pojawiały.
Bardzo istotna jest definicja, data opisu i nomenklatura Choroby Filum (EF) i syndromu Nerwowo-Czaszkowo-Kręgowego (S. NCV), jak również wcześniej opisana jednostka chorobowa, z którą mogą być mylone, czyli tak zwany: syndrom Napięcia Rdzenia (S. TM), lub syndrom Filum Terminale (S. FT), lub syndrom Napiętego Filum Terminale, lub The cord-traction syndrome lub The filum terminale syndrome lub tight filum terminale syndrome.
Również, bardzo często myli się te choroby z innymi podobnymi, takimi jak: „zakotwiczony rdzeń kręgowy” lub „Tethered cord syndrome” oraz „zakotwiczenie rdzenia poprzez ukryty rozszczep kręgosłupa” lub „Tethered spinal cord”.
Dla rozróżnienia, poniżej objaśnia się koncept oraz wszystkie te choroby, które są powiązane z FS®. Choroby te, przedstawione w terminologii hiszpańskiej oraz angielskiej, to:
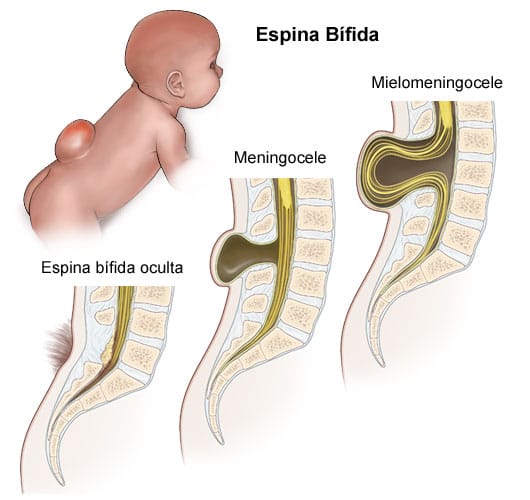
Definicja: zakotwiczenie rdzenia, lub “tethered cord syndrome” jest powiązane z występowaniem malformacji nerwowo-kręgowej, takiej jak rozszczep kręgosłupa czy przepuklina oponowo-rdzeniowa, które są widoczne zewnętrznie. Tego typu malformacje występują najczęściej w odcinku lędźwiowym kręgosłupa i kotwiczą rdzeń kręgowy, powodując urazy mechaniczne wywołane napięciem całego układu nerwowego wewnątrz czaszki oraz kręgosłupa. W wielu przypadkach obserwuje się urazy specyficzne, takie jak obniżenie migdałków móżdżku, skoliozę oraz jamistość rdzenia.
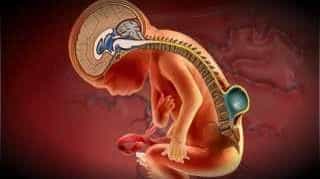
Figura 1.- Przepuklina oponowo-rdzeniowa u płodu. Na żółto zaznaczony jest rdzeń kręgowy, który powoduje silne napięcie struktur nerwowych i w konsekwencji obniżenie migdałków móżdżku do kanału kręgowego.
Opisany: przez Lichtenstein B.W. w 1940. (Lichtenstein B.W. Spinal dysraphism. Spina Bifida and myelodysplasia. Arch Neurol Psychiatry 44:792-809, 1940).
Nazwa: nazwa i definicja zostały opisane przez Yamada S. en 1996 w książce “Tethered cord Syndrome”. Shokei Yamada. American Association of Neurological Surgeons Publications Commitee”, en 1996.

Figura 2.- Fragment książki “Tethered cord Syndrome”, Shokei Yamada. American Association of Neurological Surgeons Publications Commitee”. 1996. Odpowiada wstępowi do pierwszego rozdziału książki, gdzie wskazuje się na występowanie polemiki odnośnie nazewnictwa tej patologii.
Definicja: kryte zakotwiczenie rdzenia, lub “tethered spinal cord” powiązane jest z malformacją nerwowo-kręgową, rozszczepem kręgosłupa lub przepukliną oponowo-rdzeniowa, które nie są widoczne na zewnątrz ciała. Zazwyczaj występuje w odcinku lędźwiowym kręgosłupa, i kotwiczy rdzeń kręgowy powodując urazy mechaniczne, zarówno tkanki nerwowej jak i kostnej, wywołane istniejącym napięciem. By móc zdiagnozować występowanie tej choroby, potrzebne są badania uzupełniające, takie jak RM lub TK. Może objawiać się obniżeniem migdałków móżdżku, skoliozą i/lub jamistością rdzenia.
Opisany: w 1976 roku przez Hoffman H.J. (1932-2004), neurochirurga z Toronto, który zdefiniował “tethered spinal cord” jako syndrom charakteryzujący się takimi samymi objawami obiektywnymi i subiektywnymi, jak zakotwiczenie rdzenia, jednak powiązany z rozszczepem kręgosłupa, który nie jest widoczny zewnętrznie.
Hoffman przedstawił 31 przypadków dzieci z wydłużonym rdzeniem kręgowym ze względu na ukryte zakotwiczenie rdzenia. Deficyty neurologiczne uległy poprawie po eliminacji napięcia rdzenia. Wyniki opublikował w artykule: The tethered spinal cord: its protean manifestations, diagnosis and surgical correction”. Hoffman HJ, Hendrick EB, Humphreys RP. Childs Brain. 1976;2(3):145-55.”
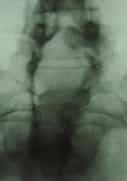
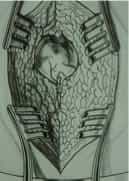
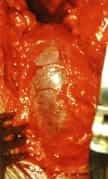
Figura 3.- Przypadek ukrytego zakotwiczenia rdzenia. Od lewej do prawej: RTG lędźwiowo-krzyżowe, z widocznym rozszczepem kręgosłupa od L4 do roztworu krzyżowego; schemat i zdjęcie operacyjne ukazujące przepuklinę oponową lędźwiowo-krzyżową, widoczną jedynie za pomocą badań uzupełniających lub in situ.
Definicja: syndrom napięcia rdzenia to syndrom kliniczny, który łączy objawy, zarówno obiektywne jak i subiektywne, związane z zaburzeniami neurologicznymi głownie dolnego poziomu rdzeniowego, połączony w możliwą skoliozą i syndromem Arnolda Chiari. Jest wynikiem napiętego filum terminale, bez jakiejkolwiek innej widocznej malformacji systemu nerwowego, łącznie z filum terminale.
Opisany: przez McKenzie, neurochirurga z Toronto. Był on pierwszym autorem, który połączył paraliż, skoliozę i napięte filum terminale, w swojej publikacji: McKenzie KG, Deward FP: “Scoliosis with paraplegia.” J Bone Joint Surg (Br) 31:162-174, 1949).
Garceau, północno-amerykański chirurg ortopeda, po raz pierwszy nadal nazwę „syndromowi napięcia rdzenia” w 1953 roku i odróżnił go od klasycznego zakotwiczenia rdzenia, w sposób jaki widoczny jest w publikacji: Garceau GJ; The filum terminale syndrome. (The cord-traction syndrome). J Bone Joint Surg (Am) 35:711-716, 1953.

Figura 4.- Z publikacji Garceau: “<<…Napięcie rdzenia połączone z rozszczepem kręgosłupa jest od lat znane neurochirurgom. W literaturze neurochirurgicznej istnieje wiele doskonałych artykułów. Syndrom ten jest powiązany ze zwykłym, ukrytym rozszczepem kręgosłupa, przepukliną oponową lub oponowo-rdzeniową, rozdwojeniem rdzenia kręgowego, wrodzoną skoliozą, syndromem Klippel-Feil, przepukliną mózgową, napiętymi pasmami włóknistymi i drzazgami kostnymi. Te wszystkie zaburzenia mogą być połączone z wrodzonymi deformacjami, zwłaszcza w kończynach dolnych. Te są najczęściej widoczne przy narodzeniu. – Trzy zaprezentowane przypadki charakteryzują się różnymi patologicznymi zaburzeniami. W pierwszym przypadku, napięte filum terminale zablokowało dogłowowe przemieszczenie się rdzenia kręgowego i ten pozostał w pozycji płodowej. W pozostałych dwóch przypadkach nie było możliwe określenie płożenia stożka rdzenia…>>
Od momentu pierwszego opisu „syndromu napięcia” w 1949 przez Mackenzie oraz definicji „syndromu trakcji rdzenia” lub “Tight filum terminale syndrome”, jako niezależnej jednostki patologicznej przez Garceau w 1953, można wyróżnić niektóre publikacje:
1. Subtle imaging findings in a case of tight filum terminale syndrome. Yeung JT, Lee CM, Fong JC. Hong Kong Med J. 2012 Jun;18(3):258-9.
2. Outcome, reoperation, and complications in 99 consecutive children operated for tight or fatty filum. Ostling LR, Bierbrauer KS, Kuntz C 4th. World Neurosurg. 2012 Jan;77(1):187-91. doi: 10.1016/j.wneu.2011.05.017. Epub 2011 Nov 19.
3. Improved symptoms and lifestyle more than 20 years after untethering surgery for primary tethered cord syndrome. Fukui J, Ohotsuka K, Asagai Y. Neurourol Urodyn. 2011 Sep;30(7):1333-7. PubMed PMID: 21626535.
4. Introductory comment: Childhood tight filum syndrome. Van Calenbergh F. Eur J Paediatr Neurol. 2012 Mar;16(2):101-2. doi: 10.1016/j.ejpn.2011.07.017. Epub 2011 Aug 31.
5. Clinical characteristics and surgical outcome in 25 cases of childhood tight filum syndrome. Cornips EM, Vereijken IM, Beuls EA, Weber JW, Soudant DL, van Rhijn LW, Callewaert PR, Vles JS. Eur J Paediatr Neurol. 2012 Mar;16(2):103-17. doi: 10.1016/j.ejpn.2011.07.002. Epub 2011 Aug 11.
6. [Congenital anomalies in the central nervous system (6) occult spinal dysraphism (other than spinal lipoma): congenital dermal sinus, tight filum terminale, neurenteric cyst, split cord malformation, and caudal regression syndrome]. Shigeta H. No Shinkei Geka. 2011 May;39(5):513-27.
7. Preoperative predictors for improvement after surgical untethering in occult tight filum terminale syndrome. Fabiano AJ, Khan MF, Rozzelle CJ, Li V. Pediatr Neurosurg. 2009;45(4):256-61. doi: 10.1159/000228983. Epub 2009 Jul 17.
8. Association of Chiari malformation type I and tethered cord syndrome: preliminary results of sectioning filum terminale. Milhorat TH, Bolognese PA, Nishikawa M, Francomano CA, McDonnell NB, Roonprapunt C, Kula RW. Surg Neurol. 2009 Jul;72(1):20-35. doi: 10.1016/j.surneu.2009.03.008. Erratum in: Surg Neurol. 2009 Nov;72(5):556.
9. Tight filum terminale syndrome in children: analysis based on positioning of the conus and absence or presence of lumbosacral lipoma. Bao N, Chen ZH, Gu S, Chen QM, Jin HM, Shi CR. Childs Nerv Syst. 2007 Oct;23(10):1129-34. Epub 2007 Jun 6.
10. A novel minimally invasive technique for spinal cord untethering. Tredway TL, Musleh W, Christie SD, Khavkin Y, Fessler RG, Curry DJ. Neurosurgery. 2007 Feb;60(2 Suppl 1):ONS70-4; discussion ONS74.
11. Miller-Dieker syndrome associated with tight filum terminale. Chen SJ, Peng SS, Kuo MF, Lee WT, Liang JS. Pediatr Neurol. 2006 Mar;34(3):228-30.
12. Occult tight filum terminale syndrome: results of surgical untethering. Albright AL. Pediatr Neurosurg. 2005 Jan-Feb;41(1):58; author reply 59-60.
13. Occult tight filum terminale syndrome: results of surgical untethering. Wehby MC, O’Hollaren PS, Abtin K, Hume JL, Richards BJ. Pediatr Neurosurg. 2004 Mar-Apr;40(2):51-7; discussion 58.
14. Management of tight filum terminale. Komagata M, Endo K, Nishiyama M, Ikegami H, Imakiire A. Minim Invasive Neurosurg. 2004 Feb;47(1):49-53.
15. Patients with urinary incontinence often benefit from surgical detethering of tight filum terminale. Selçuki M, Unlü A, Uğur HC, Soygür T, Arikan N, Selçuki D. Childs Nerv Syst. 2000 Mar;16(3):150-4; discussion 155.
16. Management of tight filum terminale syndrome with special emphasis on normal level conus medullaris (NLCM). Selçuki M, Coşkun K. Surg Neurol. 1998 Oct;50(4):318-22; discussion 322.
17. Urodynamic evaluation of tethered cord syndrome including tight filum terminale: prolonged follow-up observation after intraspinal operation. Fukui J, Kakizaki T. Urology. 1980 Nov;16(5):539-52.
18. The so-called tight filum terminale syndrome. UIHLEIN A. Minn Med. 1959 Apr;42(4):394-8.
Definicja: choroba, która charakteryzuje się bocznym skrzywieniem kręgosłupa, które wykrywa się i kontroluje jego rozwój za pomocą przeswietlenia całego kręgosłupa w pozycji frontalnej i bocznej, tzw. skoliogramu.
Rodzaje skoliozy, ze względu na przyczynę:
Definicja: skolioza idiopatyczna (ESC.I) to boczne skrzywienie kręgosłupa o nieznanej przyczynie. Częściej występuje u płci żeńskiej, zwłaszcza w okresie dojrzewania (Hipócrates IV a.c.).
Opisana: skolioza (ESC) po raz pierwszy została opisana przez Hipokratesa z Kos, w jego książce „Traktat o instrumentach redukcji”.
Nazwa: nazwę skolioza zaproponował Galeno (129-199) w połączeniu z innymi terminami, takimi jak, kifoza czy lordoza w książce „Stawy/Las Articulaciones”, w której nawiązuje do Hipokratesa i opisuje różne formy dyslokacji kręgowych.
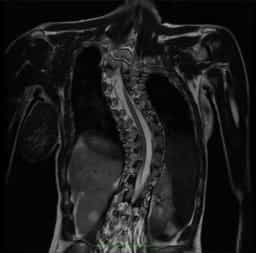
Figura 5.- RM przekrój podłużny kręgosłupa u pacjenta ze skoliozą idiopatyczną, gdzie widoczny jest kanał kręgowy i rdzeń który przechodzi od jednej wklęsłości do drugiej, wskazując na istnienie dużej siły napinającej.Teorías de la ESC.I.
Teorie ESC.I:
Gdy skrzywienie kręgosłupa, zarówno w widoku przednim jak i bocznym, powiązane jest ze znaną przyczyną: guzem, urazem, infekcją, itp.


Figura 6.- Wyjątkowy przypadek Hamiltona i Schmidta, wspomniany przez Rotha, który ukazuje ułożenie kręgosłupa oraz rdzenia kręgowego poza kanałem kręgowym u żywego pacjenta.
Definicja: syndrom Arnolda Chiari (SACH) wskazuje na przemieszczenie się dolnej części mózgu, tzw. migdałków i dolnej części móżdżku, przez otwór wielki podstawy czaszki do kanału kręgowego. Dla niektórych lekarzy, by móc zdiagnozować SACH, to przemieszczenie migdałków móżdżku (DAC) musi mieć min. 5 mm, dla innych 3 mm, a dla innych 0 mm, czyli że migdałki znajdują się na granicy otworu wielkiego, ale towarzyszy im charakterystyczny obraz kliniczny (objawy).
Opisany: w 1883 roku przez chirurga anatoma Johna Clelanda (1835-1925) z Pertshire, Szkocja. Opisał on wydłużenie robaka móżdżku, obniżenie móżdżku oraz IV komory u dziecka z wodogłowiem, przepukliną mózgu, otwartym rozszczepem kręgosłupa. W 1891 y 1896 roku, Hans Chiari wzbogacił opis nowymi przypadkami oraz klasyfikacją, a w 1894 roku, Julius Arnold przyczynił się do rozpowszechnienia tego konceptu.
Nazwany: Schwalbe i Gredig, w 1907 roku, nadali nazwę „malformacja Arnolda-Chiari”. Zgodnie z oficjalną, aktualną nomenklaturą, według Nazewnictwa i Klasyfikacji Chorób WHO (1992), stosuje się nazwę „syndrom lub choroba Arnolda Chiari (SACH).
Niektórzy autorzy nazywają tę chorobę „ malformacją Chiari”, „Chiari” lub „syndromem Chiari”. Nazwa zaproponowana przez Światową Organizację Zdrowia (WHO), w dokumencie Nazewnictwo i Klasyfikacja Chorób to „syndrom lub choroba Arnolda Chiari” (SACH). Usuniecie z nazwy „Arnolda” jest naruszeniem aktualnych reguł międzynarodowych i może prowadzić do pomyłek, czego WHO stara się unikać, ponieważ istnieją inne choroby z którymi można pomylić SACH: syndrom Chiari-Fromel, syndrom Budd-Chiari, osteologia Chiari, itp.
W Międzynarodowej Klasyfikacji Chorób WHO (Internacional Statistical Clasificación of Disecases and Relatad Health Problemas, 10th Revisión (c) Ginebra, OMS, 1992), tej chorobie przydzielona jest nazwa „syndrom lub choroba Arnolda-Chiari I” (Q07.0, CIE-10).
Występowanie: Jeden przypadek na tysiąc porodów żywych; inni autorzy wskazują na nieco mniej niż jeden procent populacji. W obu przypadkach, są to cyfry na podstawie rygorystycznych, aktualnych kryteriów DAC>3 lub 5 mm.
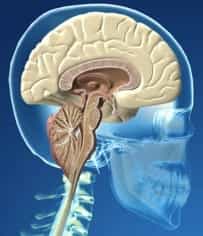
Figura 7.- Schemat przedstawiający obniżenie migdałków móżdżku i konsekwentne zwiększenie obszaru nad-móżdżkowego, w wyniku jego przemieszczenia się do kanału kręgowego w SACH.I.
Rodzaje SACH: Istnieją cztery typy klasyczne oraz dwa niedawno opisane:
I. DAC (obniżenie migdałków móżdżku) bez innej malformacji układu nerwowego.
II. DAC połączony z malformacją nerwowo-kręgową, która zakotwicza rdzeń kręgowy w kanale kręgowym.
III. DAC z przepukliną mózgową oraz innymi anomaliami mózgu.
IV. DAC z niewykształceniem lub niedorozwojem móżdżku, połączonym z niewykształceniem namiotu móżdżku.
V. Niedawno opisano występowanie przypadków z objawami typowymi dla SACH.I, jednak bez wyraźnego DAC.
VI. Niedawno został również opisany typ SACH. 1,5 z DAC oraz równoczesnym obniżeniem pnia mózgu do kanału kręgowego.
Etiologia del SAC
Obniżenie migdałków móżdżku (DAC – descenso de las amígdalas cerebelosas) może być wywołane napięciem/pociąganiem rdzenia powodowanym przez różnego rodzaju malformacje w niektórych typach SACH, oprócz SACH.I, gdzie DAC jest jedynym zaburzeniem morfologicznym. W tym przypadku istnieje kilka teorii:
Teorie konwencjonalne:
a. Hydrodynamiczna: DAC jest konsekwencją zaburzeń w przepływie płynu mózgowo-rdzeniowego (LCR).
b. Kształt czaszki: teoria o zbyt małym tylnym dole czaszki, w którym nie mieści się móżdżek, w wyniku czego przechodzi on przez otwór wielki do kanału kręgowego.
Teoria według FS®:
Uważa się, że DAC w SACH.I jest wynikiem anormalnego naciągania rdzenia kręgowego, wywołanego anormalnym filum terminale, które ściąga w dół migdałki móżdżku, oraz którego bezpośredniej anomalii nie można zaobserwować na badaniach uzupełniających.
Obraz kliniczny SACH.I może przejawiać się w zróżnicowany sposób, łącząc różne, z ponad stu typów objawów. Wśród najpowszechniejszych, według naszych obserwacji są: bóle głowy, bóle szyi, drętwienia kończyn, zaburzenia widzenia, ból kończyn, parestezje, zaburzenia czucia, zawroty głowy, zaburzenia przełykania, bóle odcinka lędźwiowego kręgosłupa, pogorszenie pamięci, zaburzenia chodu, bóle odcinka piersiowego kręgosłupa, zaburzenia równowagi, zaburzenia mowy, zaburzenia zwieraczy, bezsenność, wymioty, utraty świadomości, drżenie.
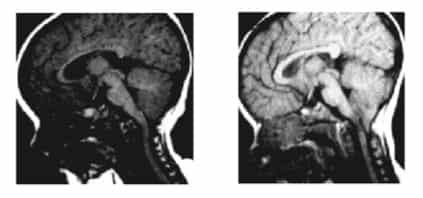
Figura 8.- RM pacjenta w 8 i 20 miesiącu życia, gdzie można zaobserwować pojawienie się DAC. Huang P.”Adquired”Chiari I malformation. J.Neurosurg 1994.
Definicja: siringomielia (SM), inaczej jamistość rdzenia, charakteryzuje się pojawianiem się cysty (jamy) w rdzeniu kręgowym, połączonej z objawami ogólnego uszkodzenia rdzenia kręgowego, a zwłaszcza zaburzenia czucia temperatury.
Opisana: siringomielia (SM) po raz pierwszy została opisana przez Estienne w jego pracy ‘La disection du corps humain’ en 1546.
Nazwana: w Paryżu, w 1824 roku, przez anatoma Charles’a Prosper Ollivier d’Angers’a (1796–1845), i opublikowana w jego “Traité de la moelle epinière et ses maladies”.
Występowanie: 84 osoby chore na milion.
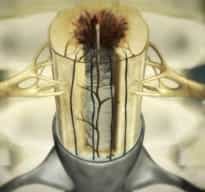
Figura 9.- Schemat ukazujący wypełnianie się jamy wewnątrz-rdzeniowej płynem tkankowym. Jama powstaje w wyniku obumierania tkanek spowodowanego niedokrwieniem wywołanym napięciem pochodzącym od filum terminale.
Rodzaje SM według etiologii:
A. Siringomielia pierwotna lub idiopatyczna (SM.I). SM.I to siringomielia lub jama wewnątrzrdzeniowa wypełniona płynem, pojawiająca się bez znanej przyczyny.
W przypadku SM.I najczęstsze objawy zaobserwowane u naszych pacjentów to: bóle kończyn, bóle szyi, zaburzenia czucia, bóle odcinka lędźwiowego kręgosłupa, bóle odcinka piersiowego kręgosłupa, bóle głowy, zaburzenia chodu, niedowłady, zaburzenia zwieraczy.
Teorie na temat etiologii SM.I:
B. Siringomielia wtórna. Według FS®, wtórna jamistość rdzenia polega na obecności jam wypełnionych płynem tkankowym, których pojawienie się wywołane jest znaną przyczyną: guzem, urazem, infekcją, itp. Prawdopodobnie wywołana jest obumieraniem tkanek rdzenia kręgowego w wyniku efektu uciskowego, inwazyjnego lub napinającego, lub kombinacji tych mechanizmów, związanego z konkretną przyczyną.
* Płaskopodstawie / Platibasia (PTB). Malformacja kostna polegająca na spłaszczeniu podstawy czaszki, które można zaobserwować na podstawie kąta podstawy lub Boogard’a. Według FS® ta deformacja kostna powstaje, gdy współistnieje plastyczna masa kostna (ze względu na niezakończony rozwój kostny lub uszkodzenie tkanki kostnej) oraz deformująca siła napinającą rdzeń w S.NCV.
* Wgłobienie obrotnika do jamy czaszki / Impresión Basilar (IB). To wgłobienie/przemieszczenie tkanek kostnych z okolicy otworu potylicznego do jamy czaszki, z równoczesnym zmniejszeniem jej objętości i zmianą kształtu podstawy czaszki na wypukły, w przeciwieństwie do normalnego (wklęsłego). Mechanizm powstania jest identyczny jak w przypadku Płaskopodstawia, gdy współistnieje plastyczna masa kostna (ze względu na niezakończony rozwój kostny lub uszkodzenie tkanki kostnej) oraz deformująca siła napinającą rdzeń w S.NCV.
* Odchylenie kręgu obrotowego / Retroceso odontoideo (RTO). Odchylenie kręgu obrotowego, to malformacja kostna polegająca na odchyleniu zęba drugiego kręgu obrotowego do tyłu, z równoczesnym przesunięciem i uciśnięciem pobliskich struktur nerwowych. Według FS®, RTO jest przejawem istnienia siły napinającej rdzeń kręgowy, która deformuje stawy, polaczenia mięśniowo-ścięgnowe oraz kości na górnym końcu kręgosłupa w S.NCV.
* Wygięcie pnia mózgu / Angulación del tronco cerebral (ATC). Wygięcie pnia mózgu polega na zmianie naturalnego kształtu pnia mózgu na wygięty, poprze obecność takich malformacji kostnych jak PTB, IB i RTO, zwłaszcza w mocno zaawansowanych przypadkach. Według FS®, odpowiada ono adaptacji przedniej zawartości dołu tylnego czaszki na deformację okolicznych struktur kostnych, spowodowanych napinaniem rdzenia w S.NCV.
* Pozostałe choroby, które mogą być wywołane napięciem rdzenia to: moczenie nocne, przepukliny dysków kręgowych, syndrom bolesnych wyrostków stawowych, syndrom Baastrup, wielokrotne dyskopatie, niewydolność krążeniowa mózgu, zaburzenia hormonalne i neuro-psychologiczne.
Definicja: syndrom Nerwowo-Czaszkowo-Kręgowy to połączenie manifestacji klinicznych oraz symptomów subiektywnych i obiektywnych, które wpływają na cały układ nerwowy, mózg, pień mózgu, rdzeń kręgowy, czaszkę i kręgosłup, jako konsekwencja anormalnie napiętego filum terminale, które pozornie wydaje się być normalne. Napięcie rdzenia może mieć przyczynę we wrodzonej, ale nie wykrywalnej wadzie filum terminale, mówimy wtedy o Chorobie Filum (Enfermedad del Filum – EF), lub może być ono nabyte, poprzez występowanie guza, urazu lub innych przyczyn, które mogą deformować kanał kręgowy i mechaniczną relację między kanałem kręgowym a połączeniem rdzeń-filum terminale.
Opisany: syndrom ten został udowodniony i opisany przez dr. Royo Salvadora, w:
Rev Neurol 1996; 24: 937-959. Siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas. Etiología común. Royo Salvador M.B.
Rev Neurol 1996; 24: 1241-1250. Impresión basilar, platibasia, retroceso odontoideo, kinking del tronco cerebral, etiología común con la siringomielia, escoliosis y malformación de Arnold-Chiari idiopáticas. Royo Salvador M.B.
Denominado: y también expuesto por el Dr. Royo Salvador en:
“Síndrome Neuro-Cráneo-Vertebral, Escoliosis, Chiari, Siringomielia. Sección del filum terminale”. XIX Jornadas de Fisioterapia de la ONCE. El Sistema craneosacro como unidad biodinámica. 6 y 7 Marzo 2009, Madrid. España
“Síndrome Neuro-Craneo-Vertebral”. IV Reunión “Chiari & Scoliosis & Syringomyelia Foundation”. 14 Octubre 2011. Palermo, Sicilia, Italia.
“Síndrome Neuro-Craneo-Vertebrale cronica, acuta, subclinica. Risultati di 400 casi operati di SEZIONE DEL FILUM TERMINALE”. V Reunión “Chiari & Scoliosis & Syringomyelia Foundation”. 12 Noviembre 2011. TRIESTE, Italia.
“Síndrome Neuro-Craneo-Vertebrale. Risultati di casi operati di SEZIONE DEL FILUM TERMINALE”. VI Reunión “Chiari & Scoliosis & Syringomyelia Foundation”. 17 Marzo 2012. ORISTANO, Italia.
“El nuevo Síndrome Neuro-Cráneo-Vertebral. La enfermedad más frecuente”. Ciclo de Charlas divulgativas CIMA (Centro Internacional Medicina Avanzada). 8 Mayo 2012. Barcelona. España.
Obraz kliniczny: syndrom Nerwowo-Czaszkowo-Kręgowy może przejawiać się klinicznie w formie jednej lub wielu chorób, które dotychczas uważane były za idiopatyczne, w większości niepołączone ze sobą, i opisane w różnych momentach w historii Medycyny. Tymi chorobami są: syndrom Arnolda Chiari I, idiopatyczna jamistość rdzenia, skolioza idiopatyczna, wgłobienie obrotnika do jamy czaszki, odchylenie kręgu obrotowego, wygięcie pnia mózgu, płaskopodstawie, niektóre zaburzenia naturalnej osi kręgosłupa (kifoza, roto-skolioza, hiper-lordoza, utrata wyrównania kręgów, itp.), niektóre zaburzenia neuro-psychologiczne, niektóre zespoły związane z niewydolnością krążeniową mózgu, niektóre dyskopatie kręgowe, niektóre przypadki syndromu bolesnych wyrostków stawowych, moczenie nocne.
Występowanie: na podstawie nowych kryteriów dla SACH.I, SM.I, ESC.I kifozy, hiper-lordozy, roto-skoliozy i innych połączonych chorób, wspólne, szacunkowe występowanie może dochodzić do 20% światowej populacji.
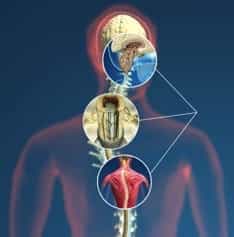
Figura 10.- Połączenie trzech chorób składa się na szeroki zakres symptomów, który może towarzyszyć S.NCV.
Definicja: Choroba Filum, jest jedną z form syndromu Nerwowo-Czaszkowo-Kręgowego, w którym wrodzone, anormalnie napięte, lecz pozornie normalne, filum terminale, powoduje napięcie w rdzeniu kręgowym oraz całym systemie nerwowym.
Nazwana: nazwa ta została przedstawiona na konferencji dla lekarzy i pacjentów “Filum System®”. Resultati in Sindrome d´Arnold Chiari, Siringomielia e Scoliosi idiopatiche”. VII Riunione “Chiari & Scoliosis & Syringomyelia Foundation”. 16 lutego 2013. Bari, Włochy.
Występowanie: na podstawie nowych kryteriów dla SACH.I, SM.I, ESC.I kifozy, hiper-lordozy, roto-skoliozy i innych połączonych chorób, oraz przewagi wrodzonego S.NCV szacunkowe występowanie może dochodzić do 20% światowej populacji.
Definicja: “Filum System®” to metoda sanitarna, która obejmuje czternaście protokołów (zastosowana u ponad 850 pacjentów, na przestrzeni 21 lat swojej historii), stosowana do leczenia Choroby Filum i sporadycznie niektórych przypadków nie wrodzonego (nabytego) syndromu Nerwowo-Czaszkowo-Kręgowego.
Nazwa: “Filum System®” podczas “Resultati in Sindrome d´Arnold Chiari, Siringomielia e Scoliosi idiopatiche”. VII Riunione “Chiari & Scoliosis & Syringomyelia Foundation”. 16 lutego 2013 roku. Bari, Włochy.
Rejestracja: zarejestrowana jako znak firmowy Nr. 3.046.839 (Marka Państwowa) i Nr. 011562725 (Marka Okręgowa). Data złożenia wniosku 26 września 2012 roku. Data zarejestrowania: 4 luty 2013 roku.
Właściciel patentu: Miguel Bautista Royo Salvador.
Od poniedziałku do czwartku: 9-18h (UTC +1)
Piątek: 9-15h (UTC +1)
Sobota, niedziela: zamknięte
+34 932 800 836
+34 932 066 406
Regulacje prawne
Informacja prawna
Pº Manuel Girona, nº 32
Barcelona, España, CP 08034
Instytut Chiari & Siringomielia & Escoliosis de Barcelona (ICSEB) spełnia wymogi Rozporządzenia UE 2016/679 (RGPD).
Zawartość tej strony web jest nieoficjalnym tłumaczeniem tekstu oryginalnego umieszczonego na stronie web po HISZPAŃSKU i jest jedynie uprzejmością Instytutu Chiari & Siringomielia & Escoliosis de Barcelona i ma na celu ułatwienie zrozumienia oryginalnego teksu osobie, która połączy się z tą stroną.






















